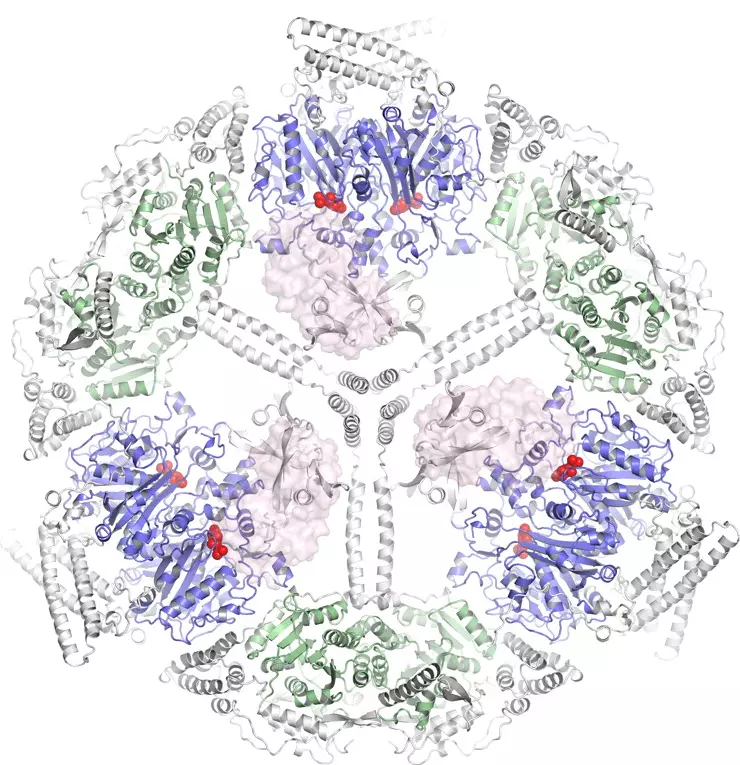
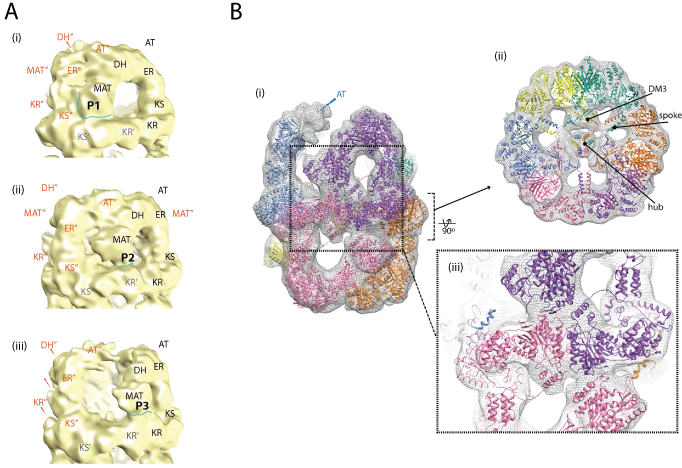Structure and function of fatty acid synthases
Important advances in the characterization of fatty acid synthase type I (FAS I) multienzyme complexes have been made during the last years by reports on X-ray crystallographic and cryo-electron microscopic (cryo-EM) structures of the bacterial, fungal and mammalian megacomplexes. In a program to structurally characterize FAS I systems, we were able to significantly contribute to this new understanding. Specifically, we structurally characterized the fungal and the bacterial FAS I, and shed light onto the acyl carrier protein (ACP) mediated substrate shuttling as well as the conformation dynamics as important inherent property of FAS I systems.
Fatty acids are carboxylic acids consisting of a polar carboxyl head group and a hydrophobic carbon tail. According to their nature, fatty acids are found between lipid and water with the carboxyl group soluble and exposed in water and the aliphatic chain elongated into the lipid phase. Most fatty acids are, however, not existing as a free fatty acid, but via ester or amide linkages as a substructure of lipids or proteins. While there is a huge variety of naturally occurring fatty acids known, a single organism uses only a few of them. In S. cerevisiae C16 (palmitic) and C18 acids (stearic) predominate. Fatty acid synthesis is performed in type I and type II systems. While in type II systems every catalytic function is provided by a separate enzyme, in type I systems, several catalytic centers are placed in a single polypeptide chain. FAS I are large multimeric proteins and generally show complex architectures.
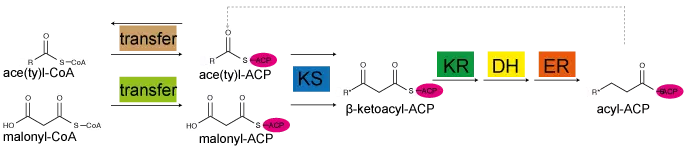
Figure. Fatty acid cycle.
Fatty acid synthesis performed in bacterial and fungal type I systems. A ketoacyl synthase (KS) domain condenses acetyl with malonyl to form β-ketobutyryl. This compound is reduced by a NADPH-dependent ketoacyl reductase (KR) to β-hydroxyacyl, dehydrated by a dehydratase (DH) to produce enoyl, and finally reduced by an enoyl reductase (ER) to the saturated acyl-product. In all these steps, the intermediates are held by ACP. Transferases load and unload ACP.
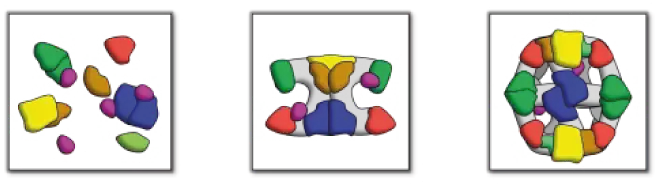
Figure. Fatty acid systems. Schematic representation of type I and type II systems. The left panel shows the separate proteins of FAS II, while complex architectural arrangements of the mammalian FAS I and fungal/bacterial FAS I are abstracted in the middle and the right panel, respectively.